Secondary structure prediction is one of the most misunderstood tools in the peptide chemist’s arsenal. For a structural biologist, an alpha-helix prediction implies a potential receptor-binding domain. For a synthetic chemist, however, a beta-sheet prediction is a red flag—a warning of “difficult sequences,” resin aggregation, and potential synthesis failure.
Peptalyzer™ utilizes a statistical, windowed approach to estimate the structural bias of a peptide sequence. This article delves into the mechanisms behind peptide secondary structure prediction, why we favor local propensity over deep learning for short peptides, and how to use these metrics to prevent aggregation on the resin.
Scan for Beta-Sheet Hotspots with Peptalyzer™
Use Peptalyzer™ to map your peptide’s local secondary structure propensity and visually flag high-risk aggregation zones (>40% sheet density) before starting your synthesis.
📘 What will you learn here?
The Basics in Peptide Secondary Structure Prediction: Propensity vs. Folding
Before interpreting your results, it is critical to distinguish between propensity and folded structure. In globular proteins, secondary structures are locked in place by a hydrophobic core and long-range tertiary interactions. Short synthetic peptides (typically <30 residues) lack this stabilizing scaffold.
Therefore, Peptalyzer™ does not predict the static 3D shape of your peptide in solution; it calculates the conformational preference of the amino acid sequence based on local thermodynamics.
- Alpha-Helix (α): A right-handed coiled rod stabilized by intrachain hydrogen bonds between the carbonyl oxygen of residue i and the amide proton of residue i+4. It requires approximately 3.6 residues per turn. High helical content often correlates with good solubility in organic solvents, provided the helix is not amphipathic (segregated hydrophobic/hydrophilic faces).
- Beta-Sheet (β): Extended polypeptide strands stabilized by interchain hydrogen bonds. In Solid-Phase Peptide Synthesis (SPPS), these are the primary driver of aggregation. The peptide chains on the resin align and stack via H-bonds, expelling solvent and causing the resin beads to collapse.
- Random Coil: Flexible, disordered regions lacking defined H-bonding patterns. From a synthesis perspective, these are ideal—they are generally well-solvated and accessible to reagents.
Propensity vs. Reality: Amino acid propensities (e.g., Alanine is a helix-former) are derived from statistical analysis of globular protein databases (PDB). They represent a chemical “preference,” not a physical guarantee. A 10-mer peptide predicted to be a helix may still behave as a random coil in water due to the high entropic cost of ordering a short chain without a hydrophobic core.
The Science: Propensity Scales and Sterics
Peptalyzer™ performs peptide secondary structure prediction based on canonical Chou-Fasman parameters, a foundational statistical method that remains highly relevant for analyzing local peptide architecture. The algorithm assigns a “conformational parameter” (P) to each of the 20 amino acids based on their frequency in specific structures. These parameters are defined only for the 20 canonical amino acids based on statistical analysis of protein structures. In Peptalyzer™, noncanonical residues are incorporated through curated residue-library mapping when a chemically defensible canonical analog exists. Depending on the residue, this may result in full support, approximation under exploratory mode, or exclusion when no valid mapping is possible. As a result, secondary structure prediction with noncanonical sequences is conditional and depends on residue compatibility. Full details of support levels, mapping rules, and strict versus exploratory behavior are provided in the noncanonical amino acids guide.
Understanding why certain residues favor specific structures allows you to read the sequence like a chemist, not just a calculator user.
- Helix Propensity (Pα):
- Strong Formers (Pα>1.2): Glu, Ala, Leu, Met. Alanine is the best helix-former because its small side chain (-CH3) is non-bulky and rigid enough to freeze the backbone angles (ϕ,ψ) into the helical range without steric clashes.
- Helix Breakers: Proline (lacks the amide proton for H-bonding and locks ϕ at -60°) and Glycine (too flexible/entropic).
- Sheet Propensity (Pβ):
- Strong Formers (Pβ>1.3): Val, Ile, Tyr, Thr. Note that Valine, Isoleucine, and Threonine are all β-branched amino acids (branching at the first carbon of the side chain).
- The Mechanism: This bulk close to the backbone creates steric clash in the tight packing of an α-helix but fits perfectly in the extended zig-zag conformation of a β-sheet.
How Peptalyzer™ Does Peptide Secondary Structure Prediction
Many simple calculators use a “global summation” approach, averaging the scores of the entire sequence. This is mathematically flawed for peptides because it allows a helix-former at the C-terminus to “cancel out” a sheet-former at the N-terminus, resulting in washed-out averages.
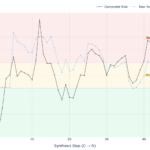
To perform peptide secondary structure prediction, Peptalyzer™ employs a Winner-Takes-All Sliding Window algorithm that separates the average character from the local risk.
The Algorithm Logic
The Window: The algorithm scans the sequence using a moving window of 6 residues. This size is chosen to approximate the minimum length required to nucleate a cooperative structure (roughly two turns of a helix or a stable beta-strand).
Local Thermodynamic Scanning: As the window moves along the sequence, Peptalyzer™ extracts the conformational parameters for Helix, Sheet, and Coil. Rather than calculating a raw average, it evaluates the relative structural competition within that specific 6-residue block.
Global Thermodynamic Potential (Overall Percentages): To determine the peptide’s general character, Peptalyzer™ calculates the total thermodynamic “pull” of each structure across the entire sequence. This nuanced approach reveals the structural conflict within the peptide (e.g., a “Chameleon” sequence might show 42% Sheet potential and 39% Helix potential), rather than artificially forcing a single winner.
The Hotspot Check (For Warning Color): Crucially, the algorithm also tracks the Maximum Sheet Density observed in any single window.
The Decision (Traffic Light): While the displayed global percentages reflect the overall peptide character, the Color Indicator is driven exclusively by the Worst-Case Window. This ensures that a single “deadly” hydrophobic knot is not mathematically hidden by a long, soluble tail.
Failsafe Behavior: For peptides shorter than 6 residues, the risk logic falls back to global propensity-based sheet estimation; in the current app UI, the Chou-Fasman graph itself is shown only for sequences of at least 7 residues.
The Classic Method vs. Peptalyzer™
To truly understand your aggregation risk, it helps to know how Peptalyzer™ improves upon the original 1970s math.
The Original Chou-Fasman Method (Raw Averages): The classic algorithm simply calculates the arithmetic mean of the raw propensity parameters (P) across a window. Because the raw parameters are centered around 1.00 (where 1.00 is neutral), averaging six strong β-sheet formers might yield a score of 1.50.
- The Flaw: A score of 1.50 is an abstract thermodynamic parameter. It tells you a sheet is likely, but it cannot be easily mapped to a standardized 0–100% risk scale for a modern user interface.
The Peptalyzer™ Method (Thermodynamic Competition): Instead of stopping at the raw average, Peptalyzer™ calculates the relative thermodynamic competition of the sequence. At any given moment, a peptide is being pulled in three directions: Helix, Sheet, or Coil. Peptalyzer™ asks: Out of the total structural pulling force in this window, what percentage is pulling toward a β-sheet?
\[\text{Sheet }\% = \left( \frac{\sum P_{\beta}}{\sum P_{\alpha} + \sum P_{\beta} + \sum P_{c}} \right) \times 100\]The Benefit for Chemists: By dividing the sheet propensity by the sum of all competing propensities, Peptalyzer™ converts abstract 1.00-level numbers into a clean 0 to 100% scale. This mathematically justifies our strict empirical risk thresholds: <30% (Safe), 30-40% (Monitor), and >40% (High Aggregation Risk).
The Chou-Fasman Parameters
The following table lists the Chou-Fasman conformational parameters (P) for the three dominant secondary structures: α-Helix, β-Sheet, and Turn. For each amino acid, these three values represent competing thermodynamic potentials—values greater than 1.00 indicate a structural preference, while values less than 1.00 indicate that the residue acts as a destabilizer or “breaker” for that specific shape.
| Amino Acid | Code | α-Helix (Pα) | β-Sheet (Pβ) | Turn (Pt) | Structural Preference |
|---|---|---|---|---|---|
| Glutamic Acid | E | 1.51 | 0.37 | 0.74 | Strong Helix |
| Methionine | M | 1.45 | 1.05 | 0.60 | Strong Helix |
| Alanine | A | 1.42 | 0.83 | 0.66 | Strong Helix |
| Leucine | L | 1.21 | 1.30 | 0.59 | Helix / Sheet |
| Lysine | K | 1.16 | 0.74 | 1.01 | Helix |
| Phenylalanine | F | 1.13 | 1.38 | 0.60 | Sheet |
| Glutamine | Q | 1.11 | 1.10 | 0.98 | Weak Helix |
| Tryptophan | W | 1.08 | 1.37 | 0.96 | Sheet |
| Isoleucine | I | 1.08 | 1.60 | 0.47 | Strong Sheet |
| Valine | V | 1.06 | 1.70 | 0.50 | Strong Sheet |
| Aspartic Acid | D | 1.01 | 0.54 | 1.46 | Turn |
| Histidine | H | 1.00 | 0.87 | 0.95 | Neutral |
| Arginine | R | 0.98 | 0.93 | 0.95 | Neutral |
| Threonine | T | 0.83 | 1.19 | 0.96 | Sheet |
| Serine | S | 0.77 | 0.75 | 1.43 | Turn |
| Cysteine | C | 0.70 | 1.19 | 1.19 | Sheet / Turn |
| Tyrosine | Y | 0.69 | 1.47 | 1.14 | Strong Sheet |
| Asparagine | N | 0.67 | 0.89 | 1.56 | Turn |
| Proline | P | 0.59 | 0.55 | 1.52 | Helix Breaker |
| Glycine | G | 0.57 | 0.75 | 1.56 | Helix Breaker |
Interpreting the Score: The Traffic Light System
Peptide secondary structure prediction is not binary. Even “helix-loving” residues contribute a small mathematical value to the Beta-sheet score (the “background noise”). For example, a simple dipeptide like Lysine-Lysine (KK) might calculate to ~25% sheet propensity purely due to mathematical normalization, despite being completely soluble.
To filter out this noise and highlight real aggregation risks, Peptalyzer™ applies the following empirical thresholds to the β-Sheet Propensity:
- < 30% (Standard Synthesis): The sequence is likely within the “safe” solubility range. The sheet score reflects background noise rather than a structured hydrophobic cluster.
- 30% – 40% (Monitor Couplings): A warning zone. The sequence contains regions with moderate aggregation potential. We recommend monitoring coupling efficiency or using double couplings.
- > 40% (Aggregation Likely): High risk. The algorithm has detected a dominant Beta-sheet motif (likely a hydrophobic cluster). This range correlates strongly with “difficult sequences” and deletion errors. Use Pseudoproline dipeptides or backbone protection here.
Why We Avoid Deep Learning for This Tool
We deliberately avoided deep-learning models (like AlphaFold) or long-range statistical methods (like GOR IV) for Peptalyzer™. Deep learning models are trained on evolutionarily related protein families and assume a globular context. They often “hallucinate” folded structures for isolated peptides because the model expects a hydrophobic core that simply isn’t there.
Windowed Chou-Fasman is chemically honest. It focuses on local interactions—the exact same interactions that cause aggregation on the SPPS resin. It doesn’t pretend to know the final fold; it alerts you to “sticky” regions.
The Chemist’s Perspective: Mitigating Aggregation Risks
For a synthetic chemist, peptide secondary structure prediction is effectively a Risk Assessment. It is important to distinguish this from kinetic difficulty: while Peptalyzer™ predicts thermodynamic aggregation (gelation) based on Chou-Fasman, the calculation for steric hindrance relies on the Krchnak et al. (1993) algorithm.
The “Beta-Sheet” Warning
“If Peptalyzer™ flags a sequence as Orange [ Monitor ] or Red [ Aggregation Likely ], it has detected a localized hotspot (>30% sheet density). You have identified a high-risk zone for aggregation. While not every beta-sheet results in a failed synthesis, this structural propensity is the primary prerequisite for “Difficult Sequences”.
- The Mechanism: Beta-sheet propensity leads to the formation of inter-chain hydrogen bond networks (C=O⋯H−N) between growing peptide chains on the solid support. This phenomenon, often called “β-sheet formation on resin,” leads to resin shrinkage and gelation. The reactive N-termini become buried in these aggregates, becoming inaccessible to incoming activated amino acids.
- The Protecting Group Factor: Remember that your peptide on the resin is not “naked”—it is covered in bulky, hydrophobic protecting groups (Trt, tBu, Pbf).
- Do PGs disrupt sheets? Generally, no. Beta-sheets are driven by backbone hydrogen bonding. Side-chain protecting groups hang off the periphery and do not block these backbone interactions.
- Do they make it worse? Often, yes. The extreme hydrophobicity of groups like Trityl (on Asn/Gln) can drive chains to cluster together to escape the polar DMF solvent, stabilizing the aggregate.
- The Symptom: Synthesis failure, usually manifesting as “deletion sequences” (missing amino acids) starting exactly after the hydrophobic cluster.
The “Beta-Sheet” Fix
- Pseudoprolines: Insert a pseudoproline dipeptide directly into the beta-sheet region. The “kink” induced by the oxazolidine ring disrupts the H-bonding network.
- Backbone Protection: Use Hmb or Dmb-protected amino acids (typically available for Gly, Ala, Val, Leu, Ile). Unlike side-chain PGs, these introduce steric bulk on the backbone nitrogen, physically blocking the inter-chain hydrogen bond.
- Pegylated Resins: Switch to ChemMatrix or Tentagel resins, which swell better in difficult sequences than Polystyrene.
The Aggregation Risk
High beta-sheet potential is the single strongest predictor of “difficult sequences” in SPPS. Unlike helices, which form intramolecular bonds, beta-sheets form intermolecular bonds. Treat any sequence with >40% predicted sheet content as a high-risk synthesis.
Limitations & “Chameleon Sequences”
Noncanonical residues: prediction depends on canonical analog mapping. Residues without defensible analogs make the Chou-Fasman output unavailable for that mode/sequence, and partial-support residues are included only in exploratory mode.
The Solvent Trap
The folding rules used by Peptalyzer™ (and all other algorithms) assume a neutral aqueous environment. However, synthesis occurs in organic solvents (DMF, NMP), and purification often uses others (ACN).
- Chameleon Sequences: A peptide may be predicted as a “Random Coil” based on its sequence, but can adopt a stable helical structure in Trifluoroethanol (TFE) or membrane-mimetic environments (SDS micelles). TFE has a low dielectric constant that strengthens electrostatic interactions, artificially stabilizing alpha-helices.
- SPPS Solvents: DMF and NMP are dipolar aprotic solvents designed to disrupt secondary structure. However, for sequences with extremely high beta-sheet propensity (e.g., amyloidogenic peptides), even DMF is insufficient to prevent aggregation.
- Thermodynamics vs. Kinetics: Peptalyzer™ calculates the thermodynamic preference for a structure. However, synthesis difficulty is also kinetic—it depends on the position relative to the resin and the reactivity of the incoming amino acid. A beta-sheet propensity at the N-terminus of a long peptide is less dangerous than one near the C-terminus.
The Length Factor
- < 10 Residues: Structural predictions are highly theoretical. Short peptides are dynamically flexible. Treat the result as a “solubility index”—hydrophobic residues will decrease solubility, regardless of the predicted “sheet” or “helix” label.
- > 20 Residues: The prediction becomes more reliable as the chain length allows for cooperative folding forces to stabilize the structure.
Peptide Secondary Structure Prediction – FAQ
Short peptides don’t fold permanently, but their propensity matters. High beta-sheet propensity drives aggregation on the resin, causing synthesis failure even if the peptide is flexible in solution.
Not necessarily. If the helix is amphipathic (hydrophobic on one side), it can form coiled-coils and aggregate. Always check the Hydropathy Plot—a “Hydrophobic Helix” is a big solubility challenge.
The PDB shows the sequence stabilized by a protein core. Peptalyzer™ sees the “naked” sequence on the resin. Without the protein scaffold, many helical sequences revert to beta-sheet aggregates.
No. Sheets are driven by backbone hydrogen bonds. Standard side-chain protecting groups (Trt, tBu) extend outward and do not block the backbone interactions that cause gelation.
Treat any sequence with >30% Beta-Sheet as a risk. If it exceeds 40%, use Pseudoprolines, backbone protection (Hmb), or elevated temperature to prevent deletion sequences.
N-methylation replaces the amide proton, making hydrogen bonding impossible – Beta-Sheet Breaker. Presence of N-methyl amino acids reduce aggregation risk.
Drastically. Peptalyzer™ assumes a neutral environment. TFE (Trifluoroethanol) artificially stabilizes helices. SDS micelles can induce folding in membrane peptides.
Yes. Heating (e.g., to 50°C) provides kinetic energy to disrupt weak beta-sheet aggregates and hydrogen bonds, often rescuing “difficult” sequences.
References
Chou, P. Y., & Fasman, G. D. (1978). Prediction of the secondary structure of proteins from their amino acid sequence. Advances in Enzymology and Related Areas of Molecular Biology, 47, 45–148.
- The definitive data monograph presenting the updated 29-protein dataset and the specific propensity values (Pα, Pβ) used in the Peptalyzer™
- DOI: 10.1002/9780470122921.ch2
Krchnak, V., Flegelova, Z., & Vagner, J. (1993). Aggregation of resin-bound peptides during solid-phase peptide synthesis. Prediction of difficult sequences. International Journal of Peptide and Protein Research, 42(5), 450–454.
- Key study distinguishing between biological folding and “resin-bound aggregation,” validating the link between hydrophobicity and synthesis failure.
- DOI: 10.1111/j.1399-3011.1993.tb00153.x
Wöhr, T., Wahl, F., Nefzi, A., Rohwedder, B., Sato, T., Sun, X., & Mutter, M. (1996). Pseudo-Prolines as a Solubilizing, Structure-Disrupting Protection Technique in Peptide Synthesis. Journal of the American Chemical Society, 118(39), 9218–9227.
- The definitive guide on using Pseudoproline dipeptides to disrupt beta-sheet aggregation in “difficult sequences.”
- DOI: 10.1021/ja961509q
Hyde, C., Johnson, T., Owen, D., Quibell, M., & Sheppard, R. C. (1994). Some ‘difficult sequences’ made easy. A study of interchain association in solid-phase peptide synthesis. International Journal of Peptide and Protein Research, 43(5), 431–440.
- Introduces Hmb backbone protection as a steric tool to physically block inter-chain hydrogen bonding.
- DOI: 10.1111/j.1399-3011.1994.tb00541.x
Kent, S. B. H. (1988). Chemical Synthesis of Peptides and Proteins. Annual Review of Biochemistry, 57(1), 957–989.
- DOI: 10.1146/annurev.bi.57.070188.004521
- The definitive review from 1988 detailing the chemical mechanisms of SPPS, including solvation effects and the physical causes of aggregation on resin.